
by Dr. Pratikkumar Rathod
ABOUT THE AUTHOR

Dr. Pratikkumar Rathod
Dr. Pratikkumar Rathod has been serving LaGuardia Community College (LaGCC) of The City University of New York (CUNY) since September 2018. Dr. Rathod is an Assistant Professor who teaches various Chemistry courses to undergraduate students of LaGCC. He earned his Ph.D. in Analytical Chemistry from the City University of New York. His expertise includes analytical chemistry, mass spectrometry, and proteomics. Dr. Rathod is actively conducting his research in the area of bioorganic chemistry and bioanalytical chemistry.
Phosphorylation: Protein phosphorylation involves catalytic addition of phosphate group from adenosine triphosphate (ATP) on serine/threonine/tyrosine residues of a protein [1]. Once a protein is phosphorylated by a kinase, the phosphorylation event can cause conformational and functional changes in the protein to either turn-on or turn-off the protein – an effective way to regulate protein functions [2]. It is estimated that about 30 percent of proteins exist in phosphorylated form at any given time in a cell, which indicates that the phosphoproteomics has vast and vital role to play in regulating various cellular processes [3].
HIV: Human Immunodeficiency Virus (HIV-1) is a causative agent of AIDS (Acquired immunodeficiency syndrome). HIV-1 specifically target host defense cells, notably CD4+T cells, and progressively destroys the ability of the host to fight HIV infection and make the host prone to other diseases. The HIV-1 virus uses various host factors for its replication; therefore, the infectivity of HIV-1 virus greatly depends on the interactions of viral components with various host factors [5].
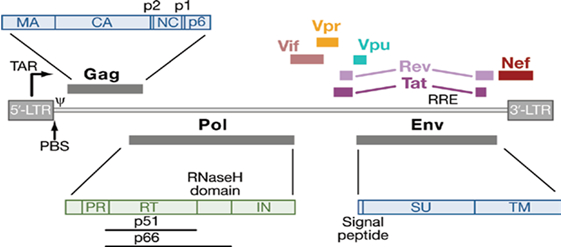
Figure 1: Structure of genome organization of HIV-1 particle [4].
Hypothesis: A few reports have been published till the date demonstrating that phosphorylation events regulate the functions of various viral proteins and play a vital role in HIV infection [6, 7], however, there is still a need for a comprehensive database. The aim of this investigation is to identify one or more novel potential phosphorylation sites across various HIV proteins and to introduce a definitive database of host-cell kinase/HIV phosphorylation substrate interactions, which may prove as a strong pillar for further investigation of anti-HIV therapeutics.
We aim to achieve the goal by following these strategic steps:
Step 1: Multiple sequence alignments of individual HIV-protein: We have constructed multiple sequence alignments using 12 different strains of HIV to identify conservation in phosphorylation sites across each HIV protein using ClustalW software. The conservation of phosphorylation sites in a protein reflects their possible biological importance in function of HIV-1 protein. In our study, we focus specifically on the Serine (Ser), Threonine (Thr) and Tyrosine (Tyr) residues that demonstrate low entropy and conservation across different strains.
Step 2: Kinase-specific phosphorylation site prediction algorithms: HIV protein sequences have been evaluated by web-based kinase-specific phosphorylation site prediction algorithms such as NetPhosK, GPS (Group-based prediction system) and KinasePhos. These algorithms not only provide prediction of potential phosphorylation sites in HIV proteins but also identify the host-kinase with prediction scores for each possible phosphorylation site.
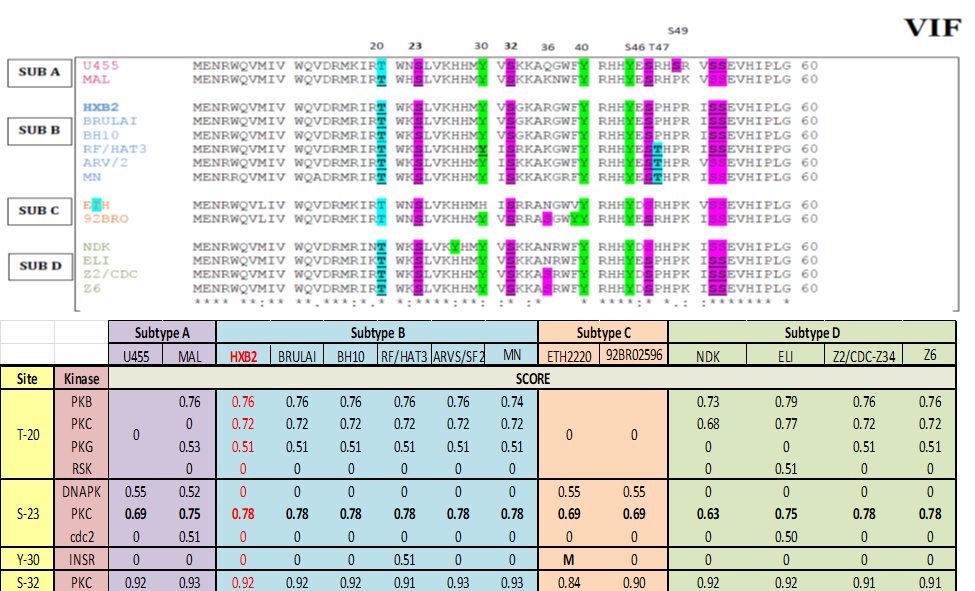
Figure 2: Vif multiple sequence alignment and bioinformatics prediction score: Top panel represent Virion infectivity factor (Vif), protein sequence from different strain is aligned together using ClustalW software identifies well-conserve phosphorylation motif in Vif protein across different strains. The residues which can be phosphorylate, such as Serine (S), Threonine (T) and Tyrosine (Y) are highlighted in purple, blue and green color respectively. Bottom panel represent scoring matrix of NetPhosK prediction scores of VIF from each strain. Notation ‘M’ represents mutation at the respected position in particular sequence. High prediction score represents high phosphorylation probability.
Step 3: In-vitro kinase assay of HIV-peptides and proteins: Libraries of short synthetic peptides derived from HIV proteins sequence containing candidate phosphorylation sites (identified from step 1 and 2) are subjected to in-vitro kinase assay with respective kinases in presence of ATP. Kinase activity is assayed using MALDI-ion trap mass spectrometer. Preliminary hits from this initial screening are then be evaluated on protein level.
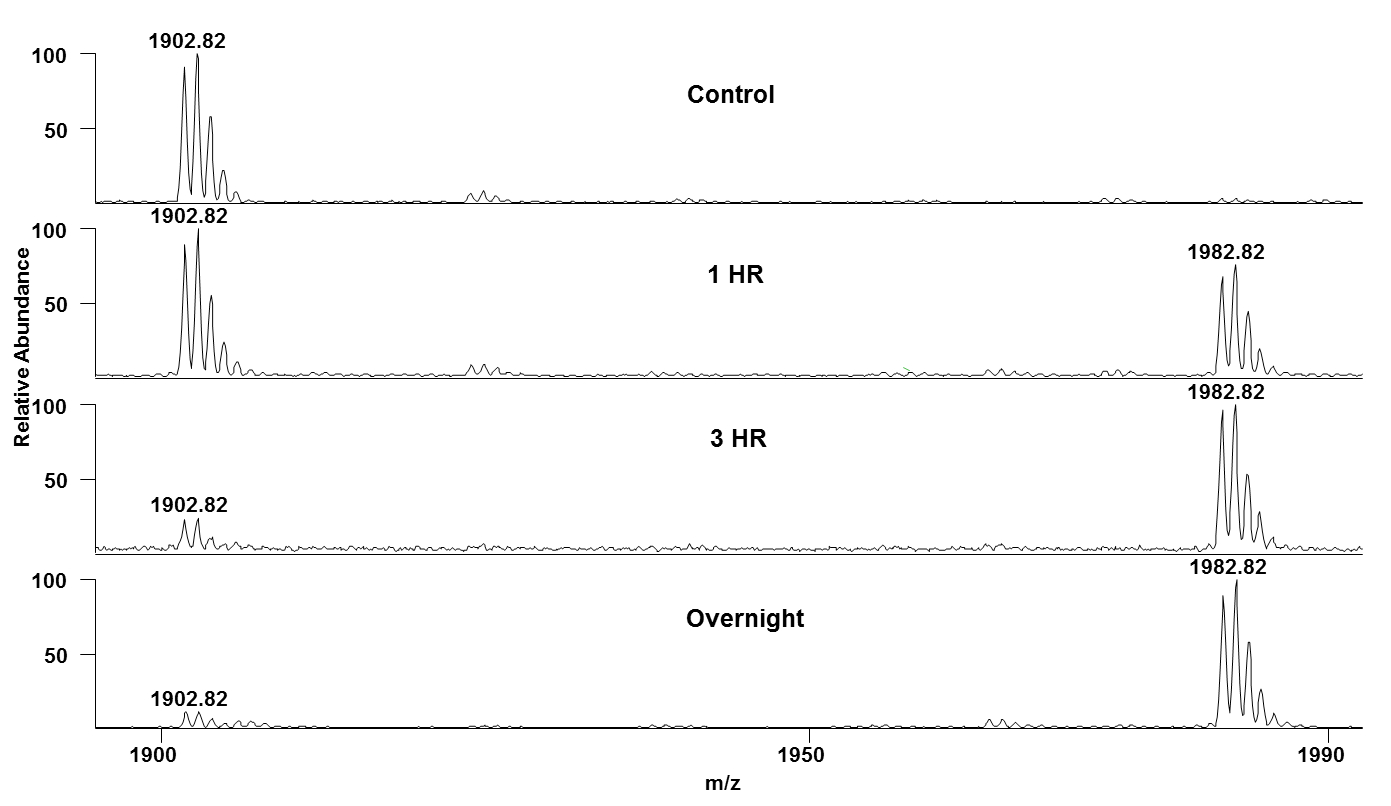
Figure 3: In-vitro kinase assay result of VIF-B-21 peptide- Mass spectra of VIF-B-21 peptide (AALITPKKIKPPLPSVTK- 1902.73Da) in presence of CDK2/Cyclin E and ATP at different time points (0hr, 1hr, 3hr, and overnight) are shown here. Time-dependent increase in the mass peak at 1982.82Da shows the CDK-dependent phosphorylation of VIF-B-21 peptides.
In this ongoing investigation, we have successfully identified and characterized more than 4 phosphorylation interactions till the date. This study will continue to confirm biological relevance of these identified in-vitro phosphorylation sites and generate comprehensive database of phosphorylation interactions between host-kinase and HIV protein sites.
[1] Mann M, Jensen ON, Nature Biotechnology, 2003; 21(3):255.
[2] Mann M, Ong SE, Grønborg M, Steen H, Jensen ON, Pandey A, Trends in Biotechnology, 2002; 20(6):261-8.
[3] Dephoure N, Gould KL, Gygi SP, Kellogg DR, Molecular Biology of the Cell, 2013; 24(5):535-42.
[4] Swanson CM, Malim MH, Cell, 2008; 133(4):742.
[5] Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ, Science, 2008; 319(5865):921-6.
[6] Barnitz RA, Wan F, Tripuraneni V, Bolton DL, Lenardo MJ, Journal of Virology, 2010; 84(13):6410-24.
[7] Leng J, Ho HP, Buzon MJ, Pereyra F, Walker BD, Xu GY, Chang EJ, Lichterfeld M, Cell Host & Microbe, 2014; 15(6):717-28.