
by Ryan Sumner and Jessica Master (Medgar Evers College, Chemistry, 2019-2020 CRSP cohort)
The work was done as a part of the CRSP program at Medgar Evers College/CUNY, under the supervision of Dr. Richard W. Denton.
This article has been published as part of the Special Edition of Ad Astra, which features the CUNY Research Scholars Program (CRSP) across The City University of New York. The issue is accessible at http://adastraletter.com/2024/crsp-special-edition/.
ABOUT THE AUTHORS

Ryan Sumner
Ryan Sumner is a dedicated and accomplished professional with experience in conflict resolution, communication facilitation, and collaborative parenting support. He is also a skilled mediator with a proven track record of minimizing legal interventions and prioritizing the child's best interests. In addition, Ryan has experience in providing tailored educational support to foster student success.
Ryan is pursuing an M.Sc. degree in Health Informatics and has already completed his B.S. in Biology from Medgar Evers College. He is proactive, adaptable, and driven, and is now seeking clinical internship opportunities to apply his diverse skill set in a healthcare setting. Ryan's ultimate goal is to work as an Informatician to improve hospital workflow and health systems. He aspires to use all the knowledge gained from his experiences to pursue a Ph.D. and educate future informaticians.

Jessica Master
Jessica Master is currently pursuing a Doctor of Pharmacy degree and a master's degree in public health at Long Island University Pharmacy. She is also interning at CVS and tutoring biology at Medgar Evers College. Jessica has been associated with Medgar Evers College since 2019, when she earned her associate degree in biology.
During her tenure at the college, Jessica has actively participated in two research projects and has maintained her position as a tutor. Her research interests revolve around the mitigation of inflammation, and she is committed to making significant contributions to the field.
Diacylglycerol (DAG) is a second messenger that activates proteins in various signaling cascades. DAG-lactones, conformationally restrained analogs of DAG, are more potent kinase C agonists. These are widely reported in the literature and are promising therapeutic targets for cancer, dementia, HIV, and multiple other disorders.
Our laboratory prepared isoxazoline (6a and 6b) as precursors to novel DAG-lactone mimetics via the microwave-assisted 3+2 cycloaddition reaction between aldoximes (7a and 7b) and 2-methylene dimethylglutarate (8). These nitrogen-containing heterocycles were eventually transformed to their respective lactone-carboxylic acid derivatives in two steps (35% for 5a and 13% for 5b). The carboxylic acid functionalities are expected to yield the desired DAG-lactones soon after borane dimethylsulfide reduction. The products isolated during synthesizing these relevant compounds were characterized using 1H- and 13C-NMR and IR spectroscopy and high-resolution mass spectrometry.
In conclusion, our project strives to synthesize novel DAG-lactone mimetics as promising therapeutic targets for life-threatening disorders. Our findings indicate a significant step towards synthesizing highly selective and potent PKC agonists, thus delivering new insights into the activation of PKC isozymes. This work has profound implications for developing new tumor and cancer research probes.
There is an urgent need to design new and more potent ligands that are molecular probes for protein kinase C (PKC) isozymes [1]. This is because PKCs are involved in signaling pathways that regulate cell growth, differentiation, apoptosis, and the promotion of tumors [2]. A potent class of PKC binding ligands prepared to probe these enzymes is the diacylglycerol (DAG)-lactones. To date, Marquez, Blumberg, and coworkers have prepared several DAG-lactones [3,4]. They identified the methylene hydroxyl groups and the lipophilic acyl chains in these molecules as essential moieties needed to increase their potency against PKC enzymes. Some potent activators of DAG-lactones include compounds 1-3. These tend to have binding affinity (Ki) values within the low nanomolar scale for PKCα and PKCε enzymes. We are actively preparing several mimetics of these potent DAG-lactones to update the present DAG-lactone pool and enhance their selectivity as probes for specific PKC isozymes.
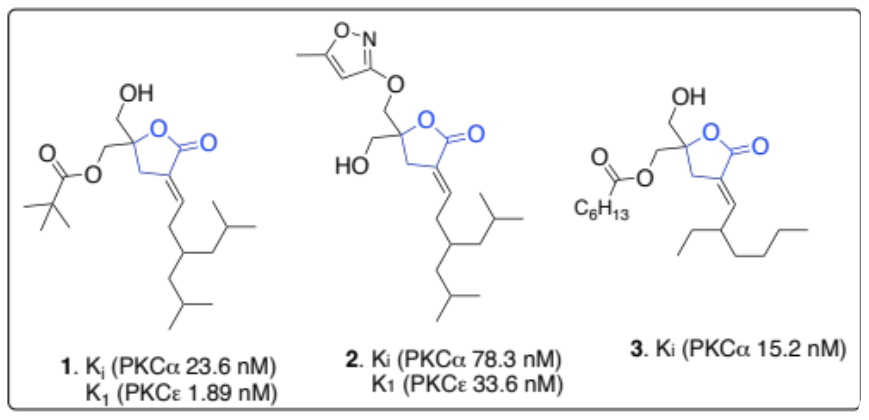
Figure 1: Representative example of some potent DAG-lactones and their inhibitory constants (Ki) for PKCα and PKCε enzymes.
Our group is skilled in applying microwave-assisted synthesis to prepare heterocyclic compounds such as isoxazolines [3]. Several groups use this protocol to synthesize nitrogen-containing heterocyclic compounds, especially isoxazolines [4]. We use microwave-assisted 2+3 cycloaddition to prepare isoxazolines en route to novel DAG-lactone mimetics. Using the docking and protein database (PDB ID:1PTR) results generated from DAG-lactone interactions with active sites on PKC-δ C1 domain [5], we designed novel DAG-lactone derivatives with slight structural modifications of the general structure A (Figure 2). These compounds possess a basic lactone moiety like compounds 1-3. The main difference in their structure was the presence of an amine (NH2/NH3+) functionality. We hypothesize that this group should form new H-bonding interactions between the amino hydrogens and the neighboring amino acids Gly 23, Leu 21, and Thr12 within the C-1 domain of the enzyme [4]. These constraints are within the predicted modes of interactions established for this enzyme.
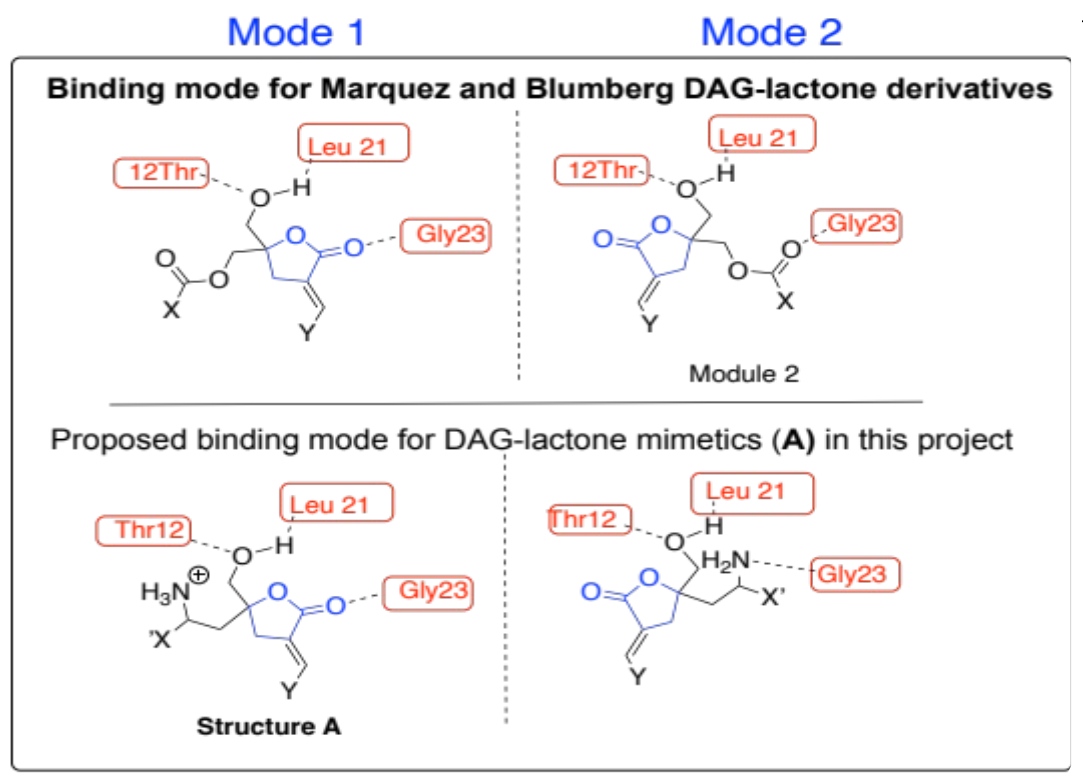
Figure 2: Illustration of the two binding modes of the DAG lactone derivatives in the C-1 domain.
Two possible DAG-lactone templates identified for synthesis were the DAG-lactone scaffolds 4a and 4b. Both compounds will be derived from the lactone carboxylic 5a and 5b, respectively (see Scheme 1).
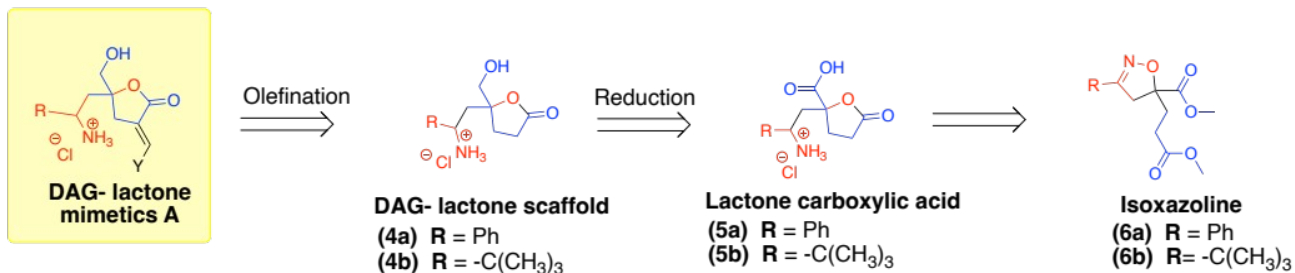
Scheme 1: Proposed synthesis of DAG lactone scaffolds 4a/4b from the isoxazolines 6a/6b.
Here, we outlined a synthetic strategy for the DAG-lactone scaffolds 4a and 4b. These compounds were proposed as suitable scaffolds for preparing new DAG-lactone derivatives as diagnostic tools for studying PKC-δ selectivity. These scaffolds were expected to be derived from the isoxazolines 6a and 6b.
Unless otherwise stated, all reactions were carried out under an air atmosphere in oven-dried glassware. Dimethyl-2-methylene glutarate (8) was synthesized from methyl acrylate and tributylphosphine under an N2 atmosphere [6]. A Perkin-Elmer Spectrum 100 FTIR Spectrometer was used to obtain IR spectra. A Bruker Advance 300 MHz FT-NMR spectrometer, using TMS or solvent peaks as a reference, was used to obtain 1H and 13C NMR spectra in CDCl3 or D2O. These spectral data assisted in the characterization of all synthesized compounds.
Thin-layer chromatography (TLC) was conducted on pre-coated silica gel plates that were visualized under UV light (254 nm) and developed with potassium permanganate stain solution or ethanolic ninhydrin in the case of amines. Microwave (MW) reactions were carried out with an Anton Par 450 monowave instrument. We reveal only the spectroscopic data for the compounds leading to the lactone carboxylic acid derivative 5a. Hence, the characterization of compounds 5a, 6a, 7a, and 9a are reported in this manuscript.
General Synthesis of Aldoximes
A mixture of oxime (52.0 mmol), hydroxylamine chloride (78.1 mmol), and a solution of 1:1 ethanol/water (30 mL) were stirred for ten minutes at room temperature. The solution was slowly added to 2 M KOH (aq) (32 mL, 62.5 mmol). The mixture was stirred for an additional 3 hours, which led to the formation of a white suspension. At this point, 1 M HCl (aq) (10.0 mL) was added to the reaction mixture, and the mixture was extracted with CH2Cl2 (2 x 20 mL). The organic layer was then dried with anhydrous Na2SO4 and evaporated to give the crude oxime product. Flash column chromatography of the crude product with 20% EtOAc/cyclohexane eluent gave the desired oximes.
Benzaldehyde oxime (7a): Yield, 97%, Clear oil, IR (neat, νmax, cm-1): 3337(O-H), 1693, 1635, 1449, 1302, 1210. 1H NMR (300 MHz, CDCl3): δ 7.38 - 7.40 (m, 3H), 7.57 – 7.60 (m, 2H), 8.17 (s, 1H), 8.70 (brd s, 1H).
General Procedure for the MW-assisted 1,3-Cycloaddition Reaction
Alkene 8 (1.2 eq), diacetoxyiodobenzene, and oxime 7a (1.0 eq) were added to a MW vial (10 mL), then 5 mL of methanol was added, and the vessel was placed in the cavity of the MW reactor. Under the reactor’s monomode setting, the vial was irradiated (150 -180°C) for 5-10 min with 1 min of hold time. The reaction was monitored by TLC until all the oxime was consumed. The solvent was evaporated via vacuum, and the residue was purified by column chromatography: silica gel (120 mesh, column packed in hexane) with 10 - 30% ethyl acetate/hexane as eluent. Each isoxazoline was afforded as a yellow oil. The reported yields were based on isolated pure product.
Methyl 5-(3-Methoxy-3-oxopropyl)-3-phenyl-4,5-dihydro-isoxazole-5-carboxylate (6a): Yield from microwave reaction, 70%. IR (neat, νmax, cm-1): 3002, 2955, 1732, 1601, 1258, 905, 759. 1H NMR (300 MHz, CDCl3): δ 2.23-2.60 (m, 4H), 3.33 (d, J = 17.3 Hz, 1H), 3.67 (s, 3H), 3.81 (s, 3H), 3.83 (d, J = 17.3 Hz, 1H), 3.36-7.45 (m, 3H), 7.62-7.65 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 28.8, 32.0, 52.0, 53.2, 88.0, 127.0, 128.0, 128.9, 156.4, 172.0, 173.0.
Synthesis of Dimethyl 2-(2-((tert-butoxycarbonyl)amino)-2-phenylethyl)-2-hydroxypentanedioate (9a)
To a solution of isoxazoline 6a (0.544 g, 1.87 mmol) in 12 mL of EtOH/THF (v:v = 1:3) under N2 atmosphere at room temperature was added di-tert-butyl-dicarbonate (0.613 g, 2.81 mmol) and NiCl2 (1.21 g, 9.34 mmol). The mixture was then stoppered and stirred for 10 minutes. The glass stopper was removed, NaBH4 (0.353 g, 9.34 mmol) was added in three portions over 30 minutes, and the flask was again stoppered. After two hours, the reaction mixture was filtered through a pad of Celite, and the filtrate was concentrated on a rotary evaporator. Flash column chromatography of the resulting crude oil using 10-30% ethyl acetate/CH2Cl2 gave the amino alcohol 9a as a white solid, dr 3:1 (0.26 g, 35%). IR (neat, νmax, cm-1): 3509, 3381, 1725. 1H NMR (300 MHz, CDCl3): δ 1.39 and 1.42 (both s, 9H), 1.90-2.52 (m, 6H), 3.56-4.02 (m, 7.5H), 4.68 (brd s, 0.25H), 5.01 (apparent t, J = 10.2 Hz, 0.75H), 5.05 (apparent t, J = 10.7 Hz, 0.25 H), 5.35 (d, J = 6.5 Hz, 0.25H), 7.22–7.35 (m, 5H). 13C NMR (75 MHz, CDCl3): δ 28.4, 28.5, 35.2, 35.5, 45.1, 45.4, 45.8, 50.4, 51.9, 53.0, 53.6, 75.0, 76.0, 79.7, 80.0, 126.3, 126.6, 127.5, 128.7, 128.8, 142.5, 142.8, 155.2, 155.4, 173.6, 175.5, 176.2. HRMS(ESI): m/z calc. for C20H29NO7Na [M+Na]+ 418.1842, found 418.1839.
Synthesis of 2-(2-Amino-2-Phenyl ethyl)-5-oxotetrahydro-furan-2-carboxylic acid (5a)
A 10-mL MW vial containing amino alcohol 9a (20.0 mg, 0.05 mmol) in 3 mL of 6 M HCl (aq) was stopped and heated in a microwave reactor at (170°C, 200 W) for 5 minutes. The reaction mixture was then transferred to a round bottom flask, concentrated by rotatory evaporation, and further lyophilized to give 5a as a white solid (14.4 mg, quantitative yield). IR (neat, νmax, cm-1): 3538-2913 (O-H carboxylic acid), 1774, 1715, 1609, 938, 7667. 1H NMR (300 MHz, D2O): δ 2.11-3.05 (m, 6H), 4.55 (dd, J = 8.4, 4.8 Hz, 0.8H), 4.99 (dd, J = 7.5, 5.7 Hz, 0.2H), 7.33-7.44 (m, 5H). 13C NMR (75 MHz, D2O): δ 27.3, 31.8, 39. 8, 52.3, 85.5, 85.9, 125.6, 127.4, 127.7, 129.0, 129.6, 135.4, 174.5, 179.6. HRMS(ESI): m/z calcd. for C13H14NO4 [M-H]- 248.0923, found 248.0927.
The microwave-assisted 3+2 cycloaddition reaction of the oximes (7a or 7b) and dimethyl methylene glutarate derivatives (8) with diacetoxyiodobenzene (DIB) produced the isoxazoline 6a and 6b in 70% and 59% respectively.
Both isoxazolines 6a and 6b were characterized using 1H and 13C NMR spectroscopy. Key resonances for the methylene (-N=C-CH2-) protons of the isoxazolidine rings appeared as AB quartets from 2.89 - 3.86 ppm with a coupling constant of 17.1 Hz [7]. The 13C NMR spectra of 6a and 6b displayed distinct resonances at 156.4 and 166.0 ppm, respectively, indicating the (-N=C-) moiety in the isoxazoline ring. The signals at 88.0 and 87.1 ppm resonated at the anticipated frequency for the tertiary carbon (C-5) in the isoxazoline ring.
The resulting isoxazolidines (6a and 6b) were reduced using anhydrous NiCl2 and NaBH4 with tert-BOC2O. This rendered the aminol 9a (35% yield) from the phenyl-derived isoxazoline 6a. Treatment of the tert-butyl-derived heterocyclic compound 6b produced the lactam-lactone derivative 10b in a meager yield of 13%. The challenges encountered in the reduction steps may be due to competing reduction and/or hydrolysis of the methyl ester groups in 6a and 6b (or their intermediates). Nevertheless, once isolated, compounds 9a and 10b were treated with 6 M HCl under microwave conditions to yield the lactone-carboxylic acid derivatives 5a and 5b, respectively. Compound 5a was characterized as a mixture of diastereomers from its 1H and 13C NMR spectral data: The methine protons (H-C-NH3+) resonated at δ 4.55 and δ 4.99 were used to determine the diastereomeric ratio (dr) of 4:1 for 5a. The carbon C-5 in the lactone of each diastereomer resonated at 85.5 and 85.9 ppm. As for compound 5b, the methine protons (H-C-NH3+) resonated at 3.46 and 3.42 ppm. Compounds 5a and 5b are expected to provide the corresponding DAG-lactone scaffolds 4a and 4b after reducing the carboxylic acid functional groups with borane dimethylsulfide complex (Scheme 2).
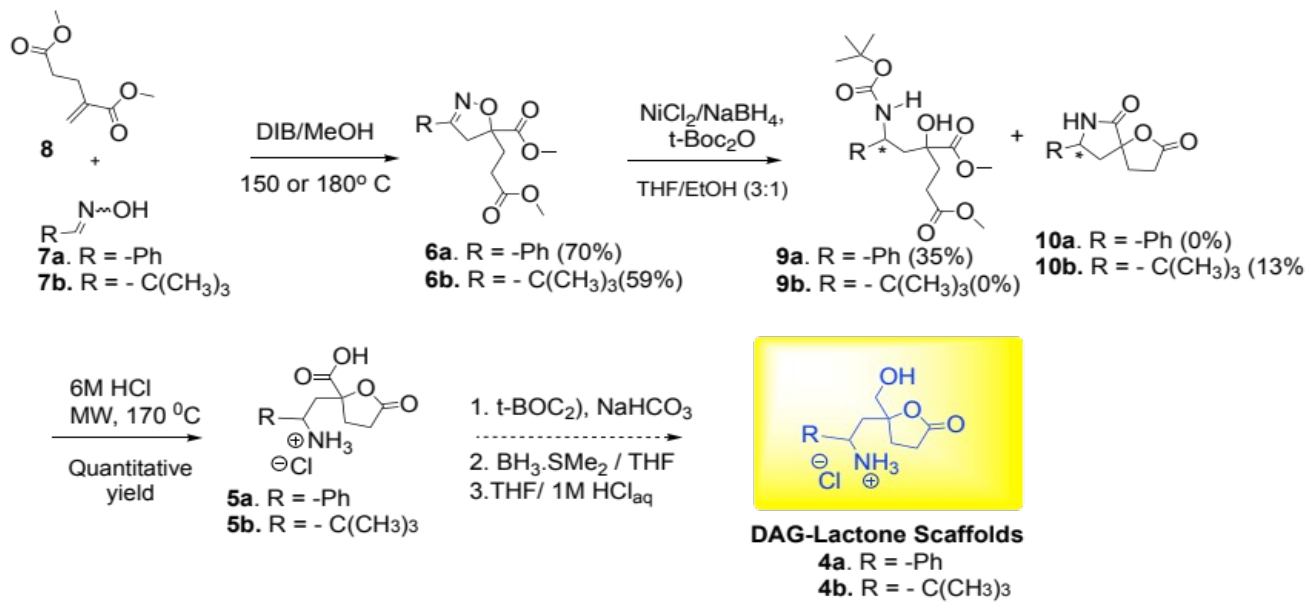
Scheme 2: Attempted synthesis of DAG lactone scaffolds 4a/4b.
In conclusion, our progress towards synthesizing two DAG-lactone scaffolds, 4a and 4b was successful. We demonstrated that it is possible to prepare DAG-lactones from isoxazoline compounds. Starting with the oximes (7a or 7b) and dimethyl methylene glutarate (8), the microwave-assisted 3+2 cycloaddition reaction occurred smoothly to provide the corresponding isoxazoline derivatives (6a and 6b) in 59-70% yield. The latter were transformed into the lactone-carboxylic acids 5a and 5b, respectively, in two steps (35% for 5a and 13% for 5b). These carboxylic acids will yield the desired DAG-lactone scaffolds soon after borane dimethylsulfide reduction. These scaffolds (4a/4b) are expected to produce novel DAG lactone derivatives as promising therapeutic probes for PKC-δ isoform.
[1] Núñez Abades, P. A., Geribaldi Doldán, N., Gómez Oliva, R., Domínguez García, S., & Castro, C. Front Cell Dev Biol, 2019, 7(39), 1-9.
[2] Tamamura, H.; Sigano, D. M.; Lewin, N. E.; Peach, M. L.; Nicklaus, M. C.; Blumberg, P. M.; Marquez, V. E. J. Med. Chem., 2004, 47(20), 4858-4864.
[3] Sumner, R.; Andromeda, X; Merrer, D. C.; Denton, R. W. Synlett 2021; 32(17), 1735-1740.
[4] (a) Chakraborty, B.; Chettri, M. S.; Luitel, G. P. J. Heterocycl. Chem., 2017, 54, 1611-1618.
(b) Al-Bogami, A. S.; Alkhathlan, H. Z.; Saleh, T. S. Asian J. Chem. 2013, 25, 6427-6433.
(c) Stassi, B.; Bougrin, K.; Soufiaoui, M. Tetrahedron Lett. 1997, 38, 8855–8858.
[5] Elhalem, E.; Bellomo, A.; Cooke, M.; Scravaglieri, A.; Pearce, L.V.; Peach, M.L.; Gandolfi Donadio, L.; Kazanietz, M.G.; Comin, M.J. J. Med. Chem, 2021, 64 (15), 11418-11431.
[6] Ayed, T. B.; El Gaied, M. M.; Amri, H. Synth. Commun. 1995, 25, 2981.
[7] Fraǵuas, R. M.; Costa, V. A.; Terra, W. C.; Aguiar, A. P.; Martins, S. J.; Campos, V. P.; Oliveira, D. F. J. Agric. Food Chem. 2020, 68, 523.